Seo Hyun Moon1-2,
Myung Haing Cho2-6,
Min Young Kim7-8 ![]()
For correspondence:- Min Kim Email: jeffmkim@jejunu.ac.kr Tel:+82647543349
Received: 6 May 2016 Accepted: 22 July 2016 Published: 30 August 2016
Citation: Moon SH, Cho MH, Kim MY. Cellular inactivation of nitric oxide induces p53-dependent apoptosis in human melanoma cells. Trop J Pharm Res 2016; 15(8):1595-1603 doi: 10.4314/tjpr.v15i8.1
© 2016 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
Purpose: To examine the role of endogenous nitric oxide (NO•) and influence of p53 status during apoptosis induced by a selective iNOS inhibitor, N-[(3-aminomethyl) benzyl] acetamidine (1400W), and/or an NO• scavenger carboxy-PTIO (c-PTIO) in two isogenic human melanoma cell lines, wild-type p53 (A375) and p53 mutant (SK mel-28) cells.
Methods: 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) and Annexin V/propidium iodide assay were used to test for antiproliferation and apoptosis, respectively. Griess and reverse transcription-polymerase chain reaction (RT-PCR) reactions were carried out to assay NO• production and the mRNA levels of inhibitors of apoptosis (IAP).
Results: c-PTIO and 1400W, alone or in combination, inhibited cell growth and promoted apoptosis via sub-G1 cell cycle arrest mediated by decrease in NO•. Apoptosis was delayed and greatly reduced in magnitude in SK mel-28 cells, underscoring the importance of p53 modulation of the response. In both cell types, apoptosis induced by iNOS inhibition and/or NO• depletion was blocked by an exogenous NO• donor, sodium nitroprusside. It was also found that inhibitors of apoptosis family (survivin, XIAP and cIAP1) were significantly depressed, which appear to play an important role in the regulation of p53-mediated apoptotic response under these conditions.
Conclusion: The data obtained provide insight into the mechanism of cell proliferation action of endogenous NO•, based on p53 status, and indicate manipulation of iNOS may offer exciting opportunities to improve the effectiveness of melanoma treatment.
Introduction
Melanoma is the most devastating skin cancer with the highest increase in incidence in recent years [1]. Melanoma originates from a malignant degenerated melanocyte, which is a highly aggressive tumor cell with poor rates of survival once it has metastasized. Melanoma is intrinsically resistant to treatments such as radiation and conventional chemotherapy, for possible reasons that involve drug-detoxifying properties of melanosomes [2], energy dependent drug efflux pumps [3], overexpression of inhibitors of apoptosis [4], altered expression of oncogenes or tumor suppressors [4] and endogenous nitric oxide (NO•) [5]. Evidently, investigating the leading drug resistant mechanisms and how to reverse or circumvent them would be critical steps in developing effective new therapies for melanoma patients.
NO• produced naturally by NO• synthase (NOS) enzymes plays a role in many different physiological processes, including vasodilation, neurotransmission, antimicrobial action, inflammation and cancer [6]. The NO• released by the constitutive enzymes neuronal NOS (nNOS) and endothelial NOS (eNOS) acts as an important signaling molecule in the cardiovascular and nervous systems [6] and NO• released by the inducible NOS (iNOS) is generated for long periods, by cells of the immune system among others, and has been shown to be principally involved in inflammatory processes and cancer formation [6].
The role for NO• in tumor biology is multi-dimensional. High level of NO• has been proposed to be cytotoxic for cancer cells. By contrast, at low level, constitutive production of endogenous NO• can promote tumor growth by inducing anti-apoptotic effects in many tumor types including melanoma [7]. Whether iNOS-derived NO• has any positive or negative effect on tumor progression, however, is still poorly understood. Numerous reports have suggested that growth and metastasis of solid tumors such as oral squamous cell carcinoma [8], breast cancer [9], melanoma [10], colon cancer [11] and gynecological malignancies [12] is correlated with an enhanced expression of iNOS. On the other hand, it has been reported that overexpression of iNOS gene can suppress tumor survival and metastasis of melanoma [13] and renal carcinoma cells [14]. From these paradoxical reports, iNOS seems to be related to anticancer as well as carcinogenesis.
The aim of the present study was to assess the inhibitory effects of endogenous NO• on the proliferation of human melanoma cells by employing a selective iNOS inhibitor 1400W, an NO• scavenger carboxy-PTIO (c-PTIO) and an NO• donor sodium nitroprusside (SNP) for modulating NO• consumption. The mechanisms of apoptotic cell death and the role of IAP family in the process were also investigated.
Methods
Cell cultures and chemicals
The choice of human tumor cells was based on p53 status. The cell line (both were a gift from Dr. G. N. Wogan, Massachusetts Institute of Technology) used were human melanoma A375 (p53 wild-type) and SK mel-28 (p53 mutant, point mutation in codon 145 of p53) cells. The cells were grown in high glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % heat-inactivated fetal bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine at 37 ºC in 5 % CO2 and cultured every few days to maintain exponential growth. Reagents and cell culture materials were purchased from the following sources: cell culture materials, Lonza (Walkersville, MD, US); Fetal Bovine Serum (FBS), PAA Laboratories (Coelbe, Germany); 1400W dihydrochloride, 2-(4-Carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (c-PTIO) and sodium nitroprusside (SNP), Sigma Chemical (St. Louis, MO, USA); N-methyl-L-arginine monoacetate (NMA) and cisplatin, CalBiochem (Salt Lake City, UT, USA).
Cell viability assay
The number of viable cells was estimated by measurement of the rate of mitochondrial metabolism of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) using a cell proliferation kit I (Roche, Indianapolis, IN, USA) according to the manufacturer’s instructions. Briefly, cells were plated at 5 × 104 cells/well in 100 μL volume in 96-well plates and were then grown for 24 h in DMEM supplemented with 10 % FBS. Different concentrations of test drugs or DMSO 0.1 % were added to the wells for the designated time points.
Cells were then incubated with 10 μL of MTT (5 mg/ml) at 37 ºC in the dark for 4 h. The tetrazolium crystals were solubilized by the addition of 100 μL of 10 % SDS in 0.01 N HCl. After overnight incubation at 37 ºC, the absorbance was measured at 550 nm using a Packard EL340 microplate reader (Bio-Tek Instruments, Winooski, VT, US). The relative percentage of cell survival was calculated by dividing the absorbance of treated cells by that of the control in each experiment. Results from the MTT assay have been used to derive the 50 % effective concentrations (EC50) of each drug to induce growth inhibition.
Apoptosis analysis
Apoptosis after treatment was quantitatively evaluated with a Becton Dickinson FACScan equipped with CellQuest software following annexin V-FITC and propidium iodide staining (Clontech Laboratories, Palo Alto, CA) following procedures reported previously [11].
Cell cycle analysis
Cells were seeded in 100-mm tissue culture dishes at a density of 2 × 106 cells/ml and were treated with 1400W and/or c-PTIO in the presence or absence of SNP for 72 h. The cells were harvested, washed twice in PBS and fixed in 70 % cold ethanol overnight. They were then suspended in 1 % BSA (bovine serum albumin)-PBS solution containing 500 μg/ml propidium iodide and 10 μg/ml RNase, incubated at 37 °C for 30 min and analyzed with a Becton Dickinson FACScan (BD Bioscience, San Jose, CA, USA). Cell fit analysis determined the percentage of cells in a specific stage of the cell cycle using CellQuest software, and expressed as a percentage of cells in the respective phases.
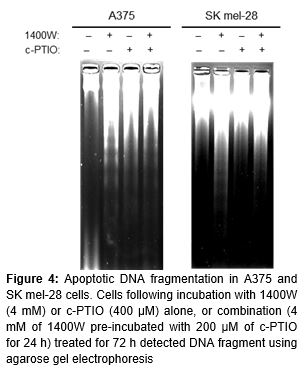
DNA fragmentation analysis
Isolation of DNA and analysis by agarose gel electrophoresis were done using a GenEluteTM mammalian genomic DNA miniprep kit (Sigma) as described [11]. Isolated DNA was suspended in TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 9.0) and quantified by absorbance at 260 nm. Fragmented DNA was loaded onto 1.8 % agarose gel containing 1 × TBE buffer and separated by electrophoresis for 2 h at 50 V, then photographed after staining with 0.5 ng/ml ethidium bromide.
Measurement of nitrite production
NO•, resulting from NOS, reacts rapidly with O2 and accumulates in the culture medium as nitrite and nitrate. NO• production was assessed by measuring nitrite in media fractions by the Griess reaction as previously described [15]. Optical density was measured using a microplate reader at 540 nm, with fresh culture media serving as the blank. Determinations were performed in triplicate. The values were obtained by comparison with standard concentrations of sodium nitrite and expressed as pmoles per 106 viable (trypan blue-excluding) cells.
Semiquantitative RT-PCR analysis
Total RNA was extracted using the Tri Reagent kit from Sigma and semiquantitative RT-PCR analysis was performed using the TOP script™ one-step RT PCR kit (Enzynomics, Daejeon, Korea) following the manufacturer’s recommended protocol. Two µg total RNA were used in each reaction. Primer sequences were as follows : for nNOS, sense 5ʹ-TTGGGGGCCTGGGATTTCTGG-3ʹ, antisense, 5ʹ-GTTGGCATGGGGGAGTGAGC-3ʹ; for iNOS, sense 5ʹ-CCAGTGACACAGGATGACCTTCAG-3ʹ, antisense 5ʹ-TGCCATTGTTGGTGGAGTAAC G-3¬ʹ; for eNOS, sense 5ʹ-CCAGCTAGCCAA AGTCACCAT-3¬ʹ, antisense 5ʹ-GTCTCGGAGC CATACAGGATT-3¬ʹ; for survivin, sense 5ʹ-GCATGGGTGCCCCGACGTTG-3¬ʹ, antisense 5ʹ-GCTCCGGCCAGAGGCCTCAA-3¬ʹ; for XIAP, sense 5ʹ-ACACCATATACCCGAGGAAC-3¬ʹ, antisense 5ʹ-CTTGCATACTGTCTTTCTGA GC3¬ʹ; for cIAP-1, sense 5ʹ-AAGTTCCTAC CCCTGTCCAATG-3¬ʹ, antisense 5ʹ-CAAGTAGA TGAGGGTAACTGGC-3¬ʹ; for cIAP-2, sense 5ʹ-CCTGTGGTTAAATCTGCCTTG-3¬ʹ, antisense 5ʹ-CAATTCGGCACCATAACTCTG-3¬ʹ; for β-actin, sense 5′-GGTCATCTTCTCGCGGTTGG CCTTGGGGT-3′, antisense 5′-CCCCAGGC ACCAGGGCGTGAT-3′. Amplified DNA was electrophoresed on 1.5% agarose gels and visualized by ethidium bromide staining.
Statistical analysis
Where necessary, statistical analysis was done by two-tailed Student’s t-test or one-way analysis of variance (ANOVA). Values of p < 0.05 were considered statistically significant.
Results
Effect of NO• on melanoma proliferation
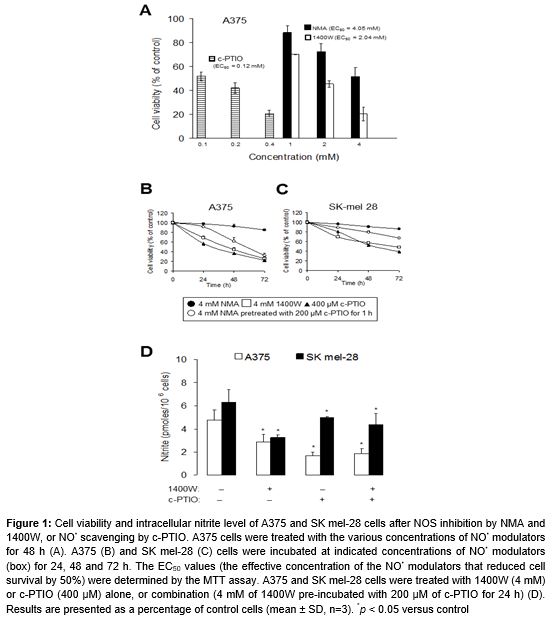
A375 cells treated with an NO• scavenger c-PTIO (0.1, 0.2 and 0.4 mM), a specific iNOS inhibitor 1400W (1, 2 and 4 mM) and the nonspecific inhibitor of NOS NMA (1, 2 and 4 mM) for 48 h responded with respect to viability, in that all treatments decreased the percentage of viable cells dose-dependently (A). The median effective concentration (EC50) with c-PTIO, 1400W and NMA to cause cell growth inhibition was found to be 0.12, 2.04 and 4.05 mM, respectively (A). We subsequently investigated the time-course of responses to 0.4 mM c-PTIO, 4 mM 1400W and 4 mM NMA, in A375 and SK mel-28 cells treated for 24, 48 and 72 h. Treatment of both cells with NOS inhibitors and NO• scavenger caused a time-dependent decrease in cell proliferation (Figures 1B and C). In comparison to A375 (p53 wild-type) cells, SK mel-28 (p53 mutant) cells were resistant to NO• modulators-induced cell death (Figures 1B and C).
Nitrite production was measured in culture media following treatment for 72 h. Significant levels of nitrite were detected in the culture medium of A375 and SK mel-28 cells (D). Cells treated with 4 mM 1400W and/or 400 μM c-PTIO for 72 h showed 1.3- to 2.8-fold decreases in nitrite level, as compared with untreated controls. p53 wild-type A375 cells produced NO• at higher concentrations than p53 mutant SK mel-28 cells (D); for example, the nitrite level in p53-WT cells (1.7 pmoles/106 cells) was thrice that of p53-null cells (5 pmoles/106 cells) after 72 h of 400 μM c-PTIO treatment.
NO• reduces loss of viability induced by cisplatin
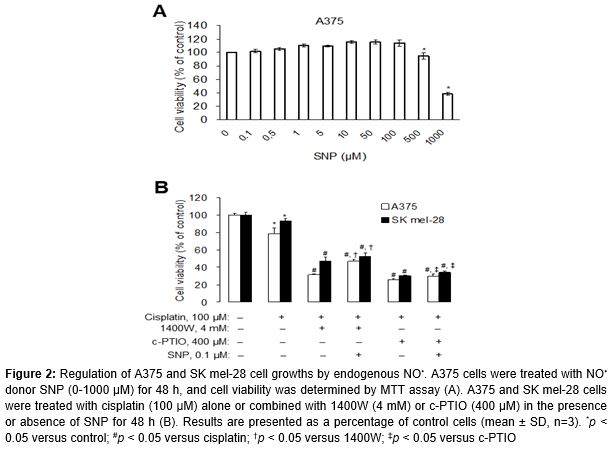
To evaluate the role of endogenous NO• in melanoma cell survival, we determined the proliferation of A375 cells at 48 h by the addition of various concentration of the NO• donor SNP. As shown in A, A375 cell proliferation was slightly increased by SNP at concentrations of 0.1-100 μM. Treatment of cells with more than 100 μM SNP caused a dose-dependent decrease in cell viability (p < 0.05).
Next, to verify whether endogenous NO• modulation regulates growth inhibition after cisplatin treatment; we incubated cells with cisplatin alone or in combination with 1400W and c-PTIO (B). Cisplatin (cis-diamminedichloroplatinum(ll)), the drug of choice for the treatment of tumors, selectively and persistently inhibits the synthesis of DNA and RNA [16].
In this study, cell viability significantly decreased by 78 and 93 % after cisplatin treatment in A375 and SK mel-28 cells, respectively (p < 0.05). When cells were co-incubated with both cisplatin and NO• modulators, viability significantly decreased by 3-folds over that which was induced by cisplatin alone in both cells (p < 0.05). Moreover, the concomitant addition of SNP, a NO• donor, with NO• modulators caused cell re-growth, which indicates that the antiproliferative effect was specific to endogenous NO• depletion (p < 0.05) (B).
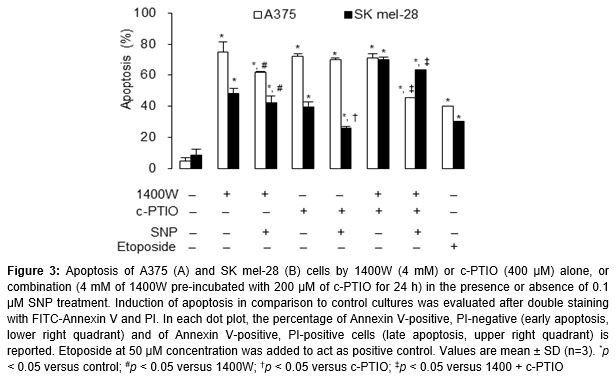
Apoptosis identified by flow cytometric analysis
To investigate the potential role of NO• in apoptosis regulation, A375 and SK mel-28 cells were treated with NO• modulators and donors SNP. shows that the iNOS inhibitor 1400W and/or the NO• scavenger c-PTIO effectively increased the cellular response to cell death, whereas those co-treated with the NO• donor SNP had an opposite effect. A stronger apoptotic response was induced in A375 (p53 wild-type) cells than in SK mel-28 (p53 mutant) cells (). Approximately 71-74 and 40-70 % of A375 and SK mel-28 cells (19- and 8-fold over control level), respectively, were apoptotic after treatment with 1400W and/or c-PTIO ().
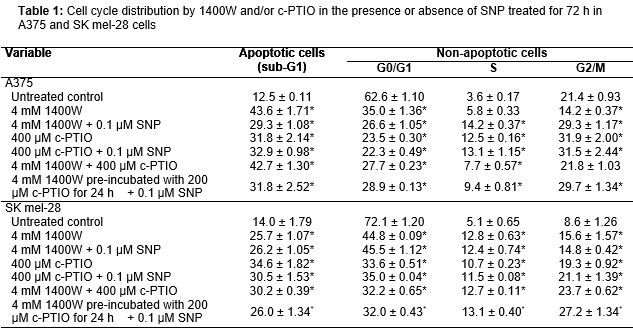
Cell cycle distribution
By FACS analysis of cell DNA content, there was a remarkable accumulation of subploid cells, sub-G1 peak, in A375 and SK mel-28 cells after treatment with NO• modulators alone or in combination with SNP for 72 h when compared with the untreated group (). Since sub-G1 peaks include early and late apoptotic cells, but also a part of necrotic cells, such large sub-G1 fractions provide strong evidence for cytotoxicity induced with 1400W and/or c-PTIO resulting in the decrease of the number of viable cells. Furthermore, the stage at which growth inhibition induced with 1400W and/or c-PTIO occurs in the A375 and SK mel-28 cell cycle progressions were determined, with cellular distribution in the different phases of the treatment (). Exposure of 1400W and/or c-PTIO resulted in a progressive and sustained accumulation of cells in the S and G2/M phases. Further, the percentage of S and G2/M phases cells increased, while those in the G1 phase decreased after treatment with 1400W and/or c-PTIO (p < 0.05) ().
Induction of internucleosomal DNA fragmen-tation induced
Since a major biochemical feature of apoptosis is fragmentation of the genomic DNA, we analyzed genomic DNA after treating the cells for 72 h. Treatments with 1400W and/or c-PTIO induced DNA ladder formation, confirming that apoptosis was a major contributor to cell death induced by these compounds ().
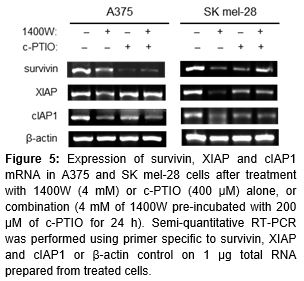
Expression of IAP family mRNA determined by semi-quantitative RT-PCR
In this study, we subsequently investigated the mechanism underlying cell death induced by endogenous NO• depletion, with a focus upon antiapoptotic activity of IAP family member. We examined expression of survivin, cIAP1 and XIAP1 mRNA. Treatment of the cells with 1400W and/or c-PTIO for 72 h led to a reduction of survivin, cIAP1 and XIAP1 in both A375 and SK mel-28 cells ().
Discussion
In recent years it has become evident that endogenous NO• has been associated with various other tumors including melanoma and high levels of tumor iNOS have been associated with tumor progression and poor survival, although the detailed molecular mechanisms involved are not clear [17]. Understanding the mechanisms involved in the modulation of endogenous NO• expression is important for the development of pharmacological strategies aimed to selectively alter the response of melanoma cells to various anticancer drugs.
Data presented here of endogenous NO• was implicated in increased melanoma cell growth; cells treated with an iNOS inhibitor 1400W and NO• scavenger c-PTIO decreased the percentage of viable cells, confirming that endogenous NO• regulation accounts for melanoma cell proliferation. Little attention had been given to these and related questions until Ekmekcioglu et al showed recently, using in 20 human metastatic melanoma tissue specimens [10], that iNOS and nitrotyrosine expression by the melanoma cells strongly correlated with poor survival in patients with stage 3 disease, suggesting a pathway whereby iNOS might contribute to enhanced tumor progression. Use of chemical NO• scavenger would nullify NO• stores by quickly reacting with any newly released NO•, whereas inhibition of NO• production by NOS does not deplete possible cellular NO• storage mechanisms [5].
The cell lines used in the present study displayed p53 function attributed in sensitivity to NO• modulators-induced cell death. In comparison to A375 (p53 wild-type) cells, SK mel-28 (p53 mutant) cells were resistant to NO• modulators-induced cell death (Figures 1B and C). The p53 protein has broad range of biological functions, including regulation of the cell cycle, apoptosis, senescence, DNA metabolism, angiogenesis, cellular differentiation, and the immune response [18]. The key role of p53 as a tumor suppressor is to block cell cycle progression and/or to induce apoptosis, in response to cellular stresses such as DNA damage. However, mutant p53 has not only led to a loss of normal function of the wild-type protein but also led to new abilities to promote cancer [19]. Mutant p53 is frequently associated with tumor progression, resistance to therapy, and poor prognosis [20].
In the present study we report that the addition of 0.1-100 μM SNP, a NO• donor, led to an increase in A375 cell growth but more than 100 μM SNP caused a dose-dependent decrease in cell viability. It suggests that low levels of NO• is involved in the maintenance of cell growth, whereas higher NO• concentrations lead to inhibition of growth, although the threshold between low and high levels of NO• is still dependent on the cell type and context within which NO• is found [6]. Similar results were observed in SK mel-28 melanoma cells (data not shown). In addition, we showed the chemoadjuvant potential of NO• modulators on cisplatin-mediated cell killing. Moreover, using SNP, cell growth returned to normal levels, confirming that NO• plays a role as a negative regulator of cisplatin-induced cytotoxicity. These results are in close agreement with earlier reported observations that NO• impairs the apoptotic function of cells and increases their resistance to cisplatin-induced cell death in human cancer cells [21]. Cisplatin induces down-regulation of Bcl-2 through proteasome emediated degradation. NO• negatively regulates this process through its ability to nitrosylate the protein and inhibit its ubiquitination. Because increased NO• production and Bcl-2 expression have been associated with several human tumors, NO• may be one of the key regulators of cell death resistance and tumor growth through S-nitrosylation [21].
The tumor suppressor p53 is activated by a variety of stress signals leading to either cell-cycle arrest or apoptosis. As expected, inhibition of endogenous NO• induced p53-dependent apoptosis in human melanoma cells. It therefore was suggested that tumor-associated NO• production promotes cancer progression by providing a selective growth advantage of p53 mutant cells. This hypothesis of a selective growth advantage of p53 mutant cells appears plausible because high concentrations of NO• and NO• metabolites, such as peroxynitrite or NO2-, cause DNA damage that in turn leads to p53-dependent apoptosis, but a sustained overproduction of NO• also is often associated with the initiation and progression of carcinogenesis [22]. Nevertheless, p53-independent processes of NO•-mediated apoptosis have also been reported [23], suggesting that other genes than p53 might also play an important role in the NO•-dependent pathways. Our data indicate that NO• plays a role as a negative regulator of p53-dependent apoptosis in melanoma cells. DNA fragmentation suggested that 1400W and/or c-PTIO induced cell death, involved a mechanism of apoptosis, this hypothesis was confirmed by flow cytometric analysis.
Since sub-G1 peaks include early and late apoptotic cells, but also a part of necrotic cells, such large sub-G1 fractions provide strong evidence for cytotoxicity induced with 1400W and/or c-PTIO resulting in the decrease of the number of viable cells. Furthermore, the stage at which growth inhibition induced by with 1400W and/or c-PTIO occurs in the A375 and SK mel-28 cell cycle progressions were determined, with cellular distribution in the different phases of treatment. 1400W and/or c-PTIO promote cell growth inhibition by inducing S and G2/M phase arrests in human melanoma cells.
Activation of the NF-κB transcription factor plays an important role in the inhibitory pathway against apoptosis [24]. The IAP family such as survivin, XIAP and cIAP1 is regulated transcriptionally by NF-κB [25]. These IAPs have been reported to directly bind and repress the activation of caspases such as caspase-3 and caspase-9, indicating that expression of IAPs under the control of NF-κB plays a pivotal role in the anti-apoptotic pathway [25]. Treatment of the cells with 1400W and/or c-PTIO for 72 h led to a reduction of survivin, cIAP1 and XIAP1 in both A375 and SK mel-28 cells, suggesting the high expression of these IAPs in human melanoma cells may act as a contributing factor to resistance by these two NO• modulators. However, our study provides some clues to elucidate the signaling pathways of the IAP genes because semi-quantitative RT-PCR can be highly variable and may not accurately reflect gene activity. Further study will be required to quantitatively measure IAP protein levels by western blotting.
Conclusion
The results of this study show that the depletion of endogenous NO• inhibits the growth of human melanoma by inducing cell cycle arrest and apoptosis via IAP inhibition, which acts as a protective factor against apoptosis in human melanoma cells. p53 wild-type A375 cells were shown to be more sensitive to apoptosis induction by NO• as compared to the p53 mutant SK mel-28 cells, suggesting that NO• is required for p53 activation. While these findings generally support the potential utility of these compounds as cancer chemopreventive or therapeutic agents, further research will be required to define more specifically the mechanisms through which they act. Recognition that NO• is a critical intermediate will facilitate progress towards that goal.
Declarations
Acknowledgement
References
Archives


News Updates